Neuroprotective effects of treadmill exercise on BDNF and PI3-K/Akt signaling pathway in the cortex of transgenic mice model of Alzheimer’s disease
Article information
Abstract
(AD). Although physical exercise and AD have received attention in the scientific literature, the mechanism through which treadmill exercise may impact the brain insulin signaling of AD has not been elucidated. This study aimed to evaluate the neuroprotective effects of treadmill exercise on apoptotic factors (Bcl-2/Bax ratio, caspase-3), HSP70, COX-2, BDNF and PI3-K/Akt signaling pathway in the cortex of NSE/hPS2m transgenic mice model of AD. Treadmill exercise ameliorated cognitive function in water maze test and significantly increased the level of Bcl-2/Bax ratio and HSP-70 in Tg-exe group compared to Tg-con group; on the other hand, it significantly decreased the expression of caspase-3 and COX-2 in Tg-exe group compared to Tg-con group. In addition, treadmill exercise significantly increased the expression of BDNF and PI3K/Akt in Tg-exe group compared to Tg-con group. Consequently, treadmill exercise improves cognitive function possibly via activating neurotrophic factor, BDNF and PI3k/Akt signaling pathway, and Aβ-induced neuronal cell death in the cortex of Tg mice was markedly suppressed following treadmill exercise. These results suggest that treadmill exercise may be beneficial in preventing or treating Alzheimer’s disease.
INTRODUCTION
Alzheimer's disease (AD) is a progressive, neurodegenerative disorder clinically characterized by the loss of memory and multiple cognitive dysfunctions [1,2]. Recently, AD is increasing rapidly as life expectancy is prolonged, and the number of patients is expected to increase to 80 million by 2040, as the ailment/disease is predicted to become a major social problem and a major health care issue all over the world [3].
The two major pathological hallmarks of AD are the extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) in the brain, yet the correct pathogenic mechanisms remain unclear. However, many recent reports and reviews have suggested that the accumulation of amyloid-β (Aβ) peptide is widely accepted as having an important role in the pathogenesis of AD. Aβ protein first deposits always develop in the neocortex, only to be followed by accumulation from hippocampus to cerebellum, leading to neuronal loss, synapses destruction, and ultimately, neuronal cell death [4,5]. Furthermore, the accumulation of Aβ protein is closely linked to a state of relative mitochondria dysfunction, hyperphosphoylation of tau, inclusion-body myositis (IBMs) and insulin resistance such as type 2 diabetes mellitus (T2D) [6-9].
More recently, clinical, experimental and epidemiological evidences have indicated that brain insulin dysfunction may significantly contribute to AD, even being referred to as “type 3 diabetes” [7,9]. Insulin is produced in the within the β-cell of the and is emerging as a major mediator of energy metabolism, especially glucose metabolism, protein synthesis and gene expression [10]. Also, insulin is abundantly distributed in the brain tissue and can be partially formed by regulating neuronal growth, synapse formation and plasticity [11,12]. However, excessive accumulation of Aβ protein in the AD brain may disrupt insulin signaling pathway by hindering the combination of insulin and insulin receptors [13], leading to a decrease in the levels of phosphati-dylinositol-3 kinase (PI3-k) and protein kinase B (Akt) activity [14,15].
The PI3-K/Akt signaling pathway is responsive to metabolic signals and environmental stress and regulates cell survival, growth, differentiation, and other homeostatic functions [16]. In particular, Akt, the serine/threonine protein kinase B, is a signaling downstream of PI3-kinase which is highly expressed in the brain and has an important role in cell survival by decreasing GSK3 (glycogen synthase kinase-3) activity, which in turn may favor the condition that induces hyperphosphoylation of tau and neuronal cell death [17-19]. In addition, Aβ protein has been shown to induce neuronal cyclooxygenase-2 (COX-2) in AD, leading to neuronal cell death and cognitive dysfunction [20]. Kotilinek et al. [21] showed that COX-2 expression is induced by Aβ protein, indicating that inhibition of COX-2 may protect Aβ-mediated suppression of memory function in AD. Therefore, reducing Aβ protein is the main target for prevention and treatment of AD by activating insulin signaling pathways and inhibiting COX-2 protein.
As mentioned above, inhibiting Aβ protein production or delaying its accumulation are of great focus, but most therapies have depended primarily on drugs so far. Pharmacological treatments could improve cognitive function for AD; however, its effect is only temporary and limited, and may produce unexpected side effects for AD. On the other hand, new integrated non-pharmacological approaches concerning/involving physical exercise can help the brain function more than approaches through medicine [22, 23]. Among the positive effects of physical exercise are the enhancement of cognition function and up-regulation of neurotrophic factors such as insulin signaling pathway (PI3-k/Akt) and brain derived neurotrophic factor (BDNF) [24-26]. However, despite the positive physiologic change observed with treadmill exercise by previous studies, treadmill exercise effects on the brain insulin signaling pathway and the neurotrophic factor have not been examined in the cortex of AD.
More interestingly, we found that NSE/hPS2m transgenic mice of AD model markedly increased the level of serum insulin, glucose and total cholesterol, suggesting that this model may have insulin resistance problem [27]. However, the mechanism linking the effect of treadmill exercise on insulin resistance is not unclear in NSE/hPS2m transgenic mice. In the present study, we used NSE/hPS2m transgenic mice model of AD to investigate whether treadmill exercise could improve cognitive function in the Morris water maze task and suppresses Aβ-induced apoptotic factor (Bcl-2/Bax ratio, caspase-3, COX-2). Moreover, we examined the neuroprotective effects of treadmill exercise on BDNF, HSP70, and PI3-K/Akt signaling in the cortex of NSE/hPS2m transgenic mice model of AD. Therefore, our data are helpful for better understanding the neuroprotective effects of treadmill exercise in the pathogenesis of AD.
METHODS AND MATERIALS
Transgenic mice
All animal experiments in this study were approved by the Institutional Animal Care and Use Committee at Korea National Sport University and by the Korea FDA. Transgenic mice, Tg-NSE/hPS2m, expressing human PS2 mutant under the control of neuron-specific enolase (NSE) were maintained in the genetic background of C57BL/6 × DBA/2 mice. The mice were maintained under controlled light and environmental condition (12 : 12 hour dark-light cycle, housed at 23 ± 1℃ with 50 % relative humidity) with food and water (Purina Mills, Seoul, Korea) made available ad libitum. Mice were carried out in an accredited Korea FDA animal facility in accordance with the AAALAC International Animal Care Policies (Accredited Unit-Korea Food and Drug Administration: Unit Number-000996).
Treadmill exercise
Tg-NSE/hPS2m mice and their control non-Tg mice at 24 months of age were divided into four groups: non-Tg-control mice (non-Tg-CON, n = 8), non-Tg- treadmill-exercised mice (non-Tg-EXE, n = 8), Tg-control-mice (Tg-CON, n = 8) and Tg- treadmill-exercised mice (Tg-EXE, n = 8). To perform the treadmill exercise, all mice were conducted at 5 m/min, 10 min/day for 5 days so that the mice became familiar with the treadmill-exercise environment. After this period, the treadmill exercise training was performed at 12 m/min, 60 min/day, 5 days/week for 3 months. However, the control group (CON) remained in their home cage throughout the course of the experiment.
Tissue preparation
After the treadmill exercise training, the five mice were anesthetized with pentobarbital sodium, with their brains were rapidly removed and their cortex separated on ice. The cortex samples were snap-frozen on dry ice and then stored at – 80 ℃ for Western blot analysis. The other three mice of each group were perfused transcardially with 50mM phosphate buffered saline followed by 4% paraformaldehyde in 0.1 M sodium phosphate buffer at pH 7.4 for immunohistochemistry.
Immunohistochemistry for COX-2 expression
Immunohistochemical analyses were performed as previously described [28]. The cyclooxygenase-2 (COX-2) in the sections (20μm-thick) were incubated overnight in the primary antibodies for COX-2 (1:200, rabbit polyclonal, Santa Cruz, CA, USA). The next day, the slides were washed in PBS three times for 5 min and pre-incubated in 2% normal donkey serum (NDS) for 15 min and then a secondary anti-body (Cy3 conjugated anti-goat for COX-2 ; 1 : 200 dilution Jackson Immunochemicals, West Grove, PA) was applied for 3 hours at room temperature. After several washes, the sections were mounted on cover slides slipped with Vectashield (Vector Laboratories, Peterborough, UK) and examined under an immune- fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
Western blot analysis
Western blot analyses were carried out as previously described [29]. Briefly, the cortex was homogenized with pro-prep protein extraction solution (Intron, Cat. No. 17081) and centrifuged at 13,000 rpm at 4℃ for 30min. Protein concentrations were measured by the protein assay kit (Bio-Rad, USA) and its protein (20 µg) were loaded with electrophoresis on a 10% polyacrylamide gel for 90 min, after which they were transferred to a polyvinylidene fluoride membrane (Immuno- Blot, PVDF membrane, Bio-Rad, CA, USA) for 1 hr at a constant voltage of 60 volts and blocked with TTBS (TBS with 0.1% Tween-20) containing 5% skim milk for 90 min. Each membrane was then separately incubated overnight at 4℃ with specific primary antibodies for PI3-k, p-PI3-k, Akt and p-Akt from Cell Signaling (dilution : 1 : 1000) and COX-2, HSP-70, BDNF and GADPH from Santa Cruz(dilution : 1 : 1000). Caspase-3, Bcl-2 and Bax from Sigma (dilution : 1 : 1000). The membranes were washed 4 times for 10 min with washing buffer and incubated for 1 hr with the secondary antibodies (HRP-conjugated goat anti-rabbit or HRP-conjugated goat anti-goat from Invitrogen). The membrane blots were then developed using ECL reagents (Santa Cruz Biotechnology, CA, USA) and the bands were captured in X-ray film. The band intensities were quantified by using a ChemiDoc XRS system (Bio-Rad, Hercules, CA, USA).
Morris water maze test
The Morris water maze tests were performed as previously described [30]. Mice were tested in a 1.5 m circular pool filled with opaque water maintained at 20℃∼22℃. The mice learned to use visual cues in the room to navigate to an escape platform (diameter 12 cm) located at a fixed position and hidden/submerged 1cm below the surface of the water. The mice were released into the pool from varying positions for a maximum of 60 sec. If it did not find the platform within 60 sec, the experimenter gently guided the mouse to the platform and let it sit on the platform for 20 sec. On each of the five acquisition days, we performed two trials with each animal, with an inter-trial interval of about five minutes. Three probe trials (60 sec) were conducted on the morning of the sixth day with the platform removed from the pool, and the crossing time in the location of the hidden platform was recorded. Escape latency, escape distance and swimming pattern of all trials were recorded with a video camera connected to a computer equipped with the SMART-CS (Panlab, Barcelona, Spain) program. To analyze the performance in the Morris water maze test, we performed two-way analysis of variance (ANOVA) across groups for the entire acquisition phase of six days.
Statistical analysis
The data were analyzed using SPSS (version 18.0, SPSS Inc., Chicago, IL, USA). All data were expressed as means ± SEM. Two-way analysis of variance (group × condition) was used to determine if there was a statistically significant interaction or main effect followed by Fisher’s LSD post hoc test for multiple comparisons. p < 0.05 was considered statistically significant.
RESULTS
Treadmill exercise improves cognitive function in the cortex of AD-transgenic mice
To investigate whether treadmill exercise affects cognitive function in NSE/PS2 transgenic mice model of AD, Morris water maze test was performed after 12 weeks of treadmill exercise. In the escape distances, there were significant differences for group (F = 20.46, p < .001), exercise (F = 18.91, p < .001), but no correlation effects were revealed according to the group or exercise (F = 1.86, p < .191, Fig. 1A). In the escape time, there were significant differences for group (F = 29.92, p < .001), exercise (F = 39.26, p < .001), but no correlation effects according to the group or exercise (F = 0.46, p < .506, Fig. 1) were revealed. In the swimming pattern, Tg-CON mice were longer than other groups, but Tg-EXE mice were able to swim shorter pattern to reach the platform than Tg-CON (Fig. 1C). Interestingly, Tg-mice showed a shorter distance and time as well as swimming pattern than Tg-control mice after the treadmill exercise. Taken together, these results reveal that treadmill exercise not only improved the cognitive function in non-Tg group, but might also alleviate the cognitive impairments of Tg mice.
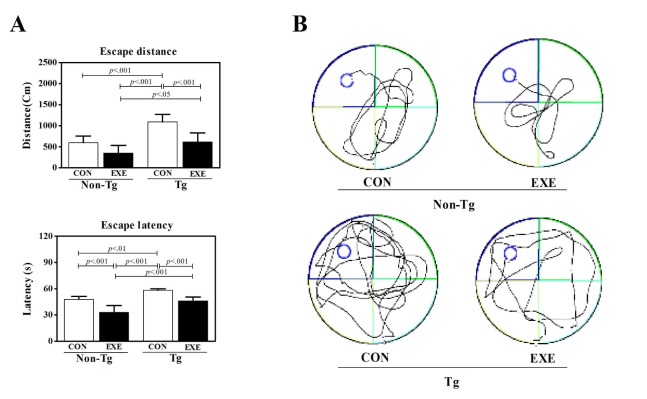
Treadmill exercise improve cognitive performance of the water maze test. (A) Patterns of escape distance, (B) escape latency. Treadmill exercise showed ameliorated ability to learn the task and displayed consistently shorter escape distances and lower escape latencies throughout testing in Tg-exe group compared to Tg-con group. (C) Patterns of swimming in mice. There was a significant difference between treadmill-exercised mice and control mice in arriving to the platform. Mean ± SEM of three trials performed using a water maze for eight mice in each of the Tg and non-Tg subgroups are shown.
Treadmill exercise influences Bcl-2/Bax ratio and caspase-3 expression in the cortex of AD-transgenic mic
To investigate whether treadmill exercise influences the activities of the pro and/or anti-apoptotic factor, western blot analyses were employed to assess the level of Bcl-2/Bax ratio and caspase-3 protein. Level of Bcl-2/Bax ratio data indicated significant effects for group (F = 10.99, p < .01), exercise (F = 132.18, p < .001), and the group × exercise interaction (F = 13.71, p < .01, Fig. 2B). The level of Bcl-2/Bax ratio in Tg-CON group was significantly lower than other groups, whereas treadmill exercise increased the level of Bcl-2/Bax ratio in Tg-EXE group. Also, the level of caspase-3 data indicated significant effects for group (F = 338.25, p < .001), exercise (F = 215.85, p < .001), but revealed no correlation effects according to the group or exercise (F = 4.27, p < .055, Fig. 2C). The caspase-3 expression in Tg-CON group was significantly higher than other groups, whereas treadmill exercise decreased the expression of caspase-3 in Tg-EXE group. Taken together, these data demonstrate that treadmill exercise improved neuronal cell death by up-regulating Bcl-2/Bax ratio and down-regulating caspase-3 proteins.

Treadmill exercise increases Bcl-2/Bax ratio and dereases caspase-3 proteins in the cortex of Tg-NSE/hPS2m mice. (A) Expression of Bcl-2/Bax ratio and caspase-3 in the cortex were analyzed by Western blot. (B) Bcl-2/Bax ratio was significantly increased in treadmill-exercised mice compared to control mice in the cortex, whereas (C) caspase-3 protein was significantly down-regulated in treadmill-exercised mice compared to control mice in the cortex. GAPDH was probed as an internal control. ap < .05, dp < .001 vs. non-Tg-control; bp < .05, ep < .001 vs. non-Tg-exercise; cp < .05, fp < .001 vs. Tg-control mice. Values are the mean ± SEM.
Treadmill exercise decreases the cyclooxygenase-2 (COX-2) protein level in the cortex of AD-transgenic mice
To investigate whether treadmill exercise influences the neuroprotection, western blot analyses were employed to assess the level of COX-2 expression. Level of COX-2 data indicated significant effects for group (F = 0.01, p < .95), exercise (F = 129.76, p < .001), and the group and exercise condition interaction (F = 66.35, p < .001, Fig. 3B). Immun-ohistochemistry revealed that Cox-2 level in cortex of Tg mice was highly enhanced, while the enhanced expression was suppressed following treadmill exercise (p < .05) (Fig. 3C). This data demonstrate that treadmill exercise improved Aβ -induced suppression of memory and synaptic plasticity via down-regulating COX-2 proteins

Treadmill exercise decrease COX-2 in the cortex of Tg-NSE/hPS2m mice. (A-C) Expression of COX-2 protein the cortex was analyzed by Western blot and immunostaining analysis. (B) Level of COX-2 protein was significantly decreased in treadmill-exercised mice compared to control mice in the cortex. GAPDH was probed as an internal control. (C) Low-level staining of COX-2 antibodies in the cortex is seen in treadmill-exercised mice. Intensive deposition of COX-2 immunoreactivity in the cortex is unique to Tg-CON mice. bp < .05, ep < .001 vs. non-Tg-exercise; cp < .05 vs. Tg-control mice. Values are mean ± SEM.
Treadmill exercise increases the BDNF protein level in the cortex of AD-transgenic mice
To investigate whether treadmill exercise influences the activities of neurogenesis maker protein, western blot analyses were employed to assess the level of BDNF expression. Level of BDNF data indicated significant effects for group (F = 59.10, p < .001), exercise (F = 16.18, p < .001), but revealed no correlation effects according to the group or exercise (F = 0.52, p < .481, Fig. 4B). This data demonstrate that treadmill exercise improved learning and memory function via up-regulating BDNF proteins.

Treadmill exercise increase BDNF, PI3-K/Akt signaling pathway and HSP70 in the cortex of Tg-NSE/hPS2m mice. (A) Expression of BDNF, p-PI3K/t-PI3K, p-Akt/t-Akt and in the cortex were analyzed by Western blot. BDNF, PI3K/Akt and HSP70 differed significantly between non-Tg and Tg mice in the cortex (B-E). Treadmill exercised mice showed a significant increase in levels of cortex BDNF (B), p-PI3K/t-PI3K ratio (C), p-Akt/t-Akt ratio (E) and HSP70 proteins (D) compared to control mice. ap < .05, dp < .001 vs. non-Tg-control; bp < .05, ep < .001 vs. non-Tg-exercise; cp < .05, fp < .001 vs. Tg-control mice. GAPDH was probed as an internal control. Values are the mean ± SEM.
Treadmill exercise enhances PI3K/Akt signaling pathway in the cortex of AD-transgenic mice
To investigate whether treadmill exercise influences the activities of insulin signaling pathway, western blot analyses were employed to assess the level of p-PI3K/t-PI3K and p-Akt/t-Akt expression. Level of p-PI3K/t-PI3K data indicated significant effects for group (F = 10.74, p < .01), exercise (F = 215.21, p < .001), but revealed no correlation effects according to the group or exercise (F = 0.29, p < .596, Fig 4C). Also, the level of p-Akt/t-Akt data indicated significant effects for group (F = 215.55, p < .001), exercise (F = 69.12, p < .001), but revealed no correlation effects according to the group or exercise (F = 3.68, p < .073, Fig. 4E). Taken together, these results reveal that treadmill exercise increases the phosphorylation of PI3-K, induces the phosphorylation of Akt, and thus, improves insulin signaling pathway in the cortex of AD-transgenic mice.
Treadmill exercise decreases the HSP-70 protein level in the cortex of AD-transgenic mice
To investigate whether treadmill exercise influences the activities of heat shock proteins, western blot analyses were employed to assess the level of HSP-70 expression. Level of HSP-70 data indicated significant effects for group (F = 128.13, p < .001), exercise (F = 110.46, p < .001), but revealed no correlation effects according to the group or exercise (F = 0.34, p < .567, Fig. 4D). This data demonstrate that treadmill exercise enhanced neuroprotection by activating HSP-70 protein.
DISCUSSION
It is widely recognized that type 2 diabetes (T2D) is a risk factor for AD, and both diseases share common pathological features including impaired glucose metabolism, insulin resistance and accumulation of Aβ protein [7,31]. Furthermore, several studies have shown that AD mice and patients have increased peripheral insulin level, indicating the impairment of insulin signaling in the AD brain. [27,32,33]. Thus, numerous epidemiological and experimental studies strongly/firmly demonstrated reducing or inhibiting Aβ protein production and improvement of insulin signaling pathway as one goal/way for the treatment of AD.
In the present study, we demonstrated that treadmill exercise could improve cognitive function and insulin sensitivity in the cortex of Tg-NSE/hPS2m mice against Aβ-induced memory loss and impaired insulin signaling pathway. Although the expression levels of Aβ protein were not measured in this study, we used Tg-NSE/hPS2m mice model, a known increased expression of Aβ protein in the brain. In our previous studies, we found that Tg-mice show significant increase in Aβ protein, which leads to cognitive impairment and neuronal cell death [27,29,30,34]. As expected, in the present study, we found that Tg-mice show cognitive impairment, including reduction of escape distance and latencies and markedly increased neuronal cell death in the cortex of Tg mice. Therefore, we assume that cognitive deficits and the increased levels of apoptotic induce factor could also be responsible for the Aβ depositions. Although the underlying mechanisms implicated in the treadmill exercise-induced improvement of cognitive function are not completely understood, it is reasonable to postulate that decrease in Aβ deposition, neuronal cell death and increase in insulin signaling pathway after treadmill exercise may be related to the cognitive function.
Many proteins have been reported to be associated with apoptosis under physiological states, in which Bcl-2 and Bax protein are demonstrated to play a key role in neuronal cell death or survival [35]. The present study showed that cortex Bcl-2 protein was decreased and Bax protein was increased in Tg mice; however, treadmill exercise up-regulated the expression of Bcl-2 and down-regulated the expression of Bax in the cortex of Tg mice compared to Tg control mice.
According to our results, the level of Bcl-2/Bax ratio was increased after treadmill exercise. In addition, caspase-3, a pro-apoptosis factor, is activated during the last step of apoptosis and could be decreased by the Bcl-2 and increased by the Bax protein [36]. We found that the level of caspase-3 proteins were increased in the cortex of Tg mice compared to non-Tg mice while treadmill exercise induced down-regulation of the caspase-3 in the cortex of Tg mice, which was consistent with previous reports [27,37,38]. Furthermore, cyclooxygenase-2 (COX-2) contributes to synaptic plasticity and memory function whereas when overexpressed it has been associate with neurotoxicity in AD [39]. Kotilinek et al. [21] and Cakala et al. [40] demonstrated that AD animals exhibited Aβ-induced neuronal cell death and cognitive dysfunction via the activation of COX-2. In this study, Tg mice increased Aβ-induced neuronal cell death showing activation of COX-2 when compared to those of non-Tg mice; however, treadmill exercise significantly reduces the level of COX-2 immunoreactivity and Western blot in the cortex of Tg mice. These findings demonstrated that treadmill exercise might attenuate neuronal cell death in the cortex of AD by increasing Bcl-2/Bax ratio and reducing caspase-3 and COX-2 protein level.
Brain-derived neurotrophic factor (BDNF) is involved in neurogenesis and memory function, including neuronal connectivity, synaptic development and plasticity; in addition, it enhances the insulin sensitivity in patients with type 2 diabetes (T2D) [41,42]. Notably, it is well known that BDNF is widely expressed in the rodent and the human brain; it is especially abundant in the hippocampus, cerebral cortex and cerebellum [43]. However, increasing/growing evidence demonstrated that BDNF protein was decreased in the animal and patients with AD and T2D [42,44]. Several studies have shown that physical activity induced up-regulating of the BDNF protein in the hippocampus and cortex and therefore, may play a role in improving cognitive function [27,45]. Moreover, Pedersen et al. [46] suggested that BDNF is likely to mediate some of the positive effects of exercise with regard to the protection against memory dysfunction and T2D. Consistent with these finding, our present study demonstrated that the expression of BDNF proteins was decreased in the cortex of Tg mice, whereas treadmill exercise induced up-regulation of the BDNF in the cortex of Tg mice. Thus, increased expression of BDNF may also be involved in the neuroprotective and insulin sensitivity by treadmill exercise.
Growing evidence suggested that insulin function and impaired signaling pathway could be common hallmark links between T2D and AD. Indeed, insulin signaling is transduced through the PI3-k/Akt pathway and plays significant roles in the regulation of glucose metabolism in the periphery tissue as in brain function. Interestingly, the role of PI3-k/Akt pathway in the brain function has recently received much attention such as synaptic plastic, cognitive function, neuronal growth and survival [47]. Previous studies reported that AD-induced cognitive dysfunction has been postulated to be caused by impaired insulin signaling in the brain [7,47]. In addition, as presented in a study by Wang et al. [48], insulin-deficient diabetes in AD mice model deteriorated cognitive function and the accumulation of Aβ protein characterized by impaired insulin signaling pathway. Further, we found that Tg mice show decreased PI3-K/Akt signaling pathway in the cortex; however, treadmill exercise increase PI3-K/Akt signaling pathway in the cortex of Tg mice.
Heat shock proteins (HSPs), in particular HSP70, are ubiquitously expressed intracellular proteins that protect proteins and could suppress the progression of AD [49-51]. In a previous study, Magrane et al. [52] suggested that neuroprotective by Akt may be regulated through downstream factor of HSP-70 expression, indicating that deficiency of HSP70 is possibly caused by the Aβ-induced down-regulation of Akt protein. The present study showed that the level of HSP70 was decreased in the cortex of Tg mice compared to non Tg mice; however, treadmill exercise could increase HSP70 protein which is in accordance with previous study [27,37,38]. Therefore, these results demonstrated that the activation of PI3-K/Akt signaling and HSP70 might ameliorate the cognitive dysfunction in the cortex of AD.
CONCLUSION
In conclusion, we investigated the neuroprotective effect of treadmill exercise against Aβ-induced cognitive dysfunction in Tg-NSE/hPS2m mice model. Treadmill exercise significantly improved Aβ-induced cognitive dysfunction via elevation of BDNF, PI3-k/Akt pathway and HSP70. Also, treadmill exercise enhanced the neuronal cell survival, elevated Bcl-2/Bax ratio, and inhibited caspase-3 and COX-2 activity. Taken together, these results demonstrated that neuronal cell death and impaired insulin signaling pathway associated with Aβ protein may be partially decreased after treadmill exercise in the cortex of AD mice.
Acknowledgements
We thank the animal technicians Dong-H, Choi. Yo-W, Choi. In addition, we thank Seok-M, Hong for directing the animal facility at the Korea National Sport University. This study was supported by Sol grants to professor. Joon Y. Cho from the Korean Society of Sports Medicine (2012-2013).