Effects of long-term resistance exercise training on autophagy in rat skeletal muscle of chloroquine-induced sporadic inclusion body myositis
Article information
Abstract
Purpose
We examined whether resistance exercise training restores impaired autophagy functions caused by Chloroquine (CQ)-induced Sporadic Inclusion Body Myositis (sIBM) in rat skeletal muscle.
Methods
Male wistar rats were randomly assigned into three groups: Sham (n = 6), CQ (n = 6), and CQ + Exercise (CE, n = 6). To create a rat model of sIBM, rats in the CQ and CE group were intraperitoneally injected with CQ 5 days a week for 16 weeks. Rats in the CE group performed resistance exercise training 3 times a week for 8 weeks in conjunction with CQ starting from week 9 to week 16. During the training period, maximal carrying load, body weight, muscle weight, and relative muscle weight were measured. Autophagy responses were examined by measuring specific markers.
Results
While maximal carrying capacity for resistance exercise training was dramatically increased in the CE group, no significant changes occurred in the skeletal muscle weight as well as in the relative muscle weight of CE compared to the other groups. CQ treatment caused significant increases in the levels of Beclin-1 and p62, and decreases in the levels of LAMP-2 proteins. Interestingly, no significant differences in the LC3-II/I ratio or the LC3-II protein levels were observed. Although CQ-treatment groups suppressed the levels of the potent autophagy inducer, BNIP3, p62 levels were decreased in only the CE group.
Conclusion
Our findings demonstrate that sIBM induced by CQ treatment results in muscle degeneration via impaired autophagy and that resistance exercise training improves movable loading activity. Finally, regular exercise training may provide protection against sIBM by enhancing the autophagy flux through p62 protein.
INTRODUCTION
Sporadic Inclusion Body Myositis (sIBM) is the most common muscle degenerative disease in elderly populations, with a clinical feature of progressive muscle weakness and severe muscle atrophy [1–3]. The remarkable pathologic features of sIBM present as vacuolated muscle fibers, accumulations of abnormal proteins that are similar to the pathogens of Alzheimer’s and Parkinson’s diseases in the brain [4–7]. Although the exact mechanisms of sIBM remain unclear, strong evidence suggests that the primary cause could be endoplasmic reticulum (ER) stress, unfolded protein response, lysosomal inhibition, and dysfunction of protein degradation systems including autophagy [8–10]. Unfortunately, sIBM patients do not respond well to pharmacologic treatments. As a result, exercise has been recommended as a safe method of therapy with no signs of increased muscle inflammation [11–13].
Autophagy is described as a major catabolic process of long-lived bulk proteins and organelles via lysosome-dependent pathways. Following their degradative products, these proteins are recycled during the turnover of intracellular proteins [14]. In all types of autophagy (macro-, micro-, and chaperone-mediated), one prominent feature is the formation of double-membrane vesicles, termed autophagosomes, which eliminate abnormal proteins as well as damaged organelles including mitochondria and transfer them to the lysosome where they are degraded [15]. Basically, autophagy occurs constitutively at lower basal levels in all eukaryotic cells and tissues and is strongly induced by nutrient starvation. However, it is also substantially induced in response to various stressors and signals [16,17]. To activate autophagy, the formation of autophagosomes originating from the control of autophagy-related (Atg) proteins is needed. These Atg proteins modulate autophagy flux, higher rates of initiation and the resolution of autophagic events. The gene profiles emphasize the physiological functions of skeletal muscles in response to exercise [18–21]. Importantly, exercise-induced autophagy is required to maintain skeletal muscle mass and contribute to improving glucose metabolism [22–25]. However, aberrant autophagy flux is detrimental for muscle health and leads to muscle atrophy and degeneration, while sufficient autophagy flux helps maintain healthy myofibers [26]. Accordingly, an optimal level of autophagy is essential in fine-tuning skeletal muscle homeostasis in the physiological state as well as in pathological conditions [27].
Chloroquine (CQ) is a lysosomotropic agent that prevents endosomal acidification. The accumulation of CQ in lysosomes causes an increase in lysosomal pH, inhibiting the activities of lysosomal proteases and eventually diminishing autophagy flux [28]. Furthermore, the long-term effects of CQ treatment in rats cause myopathy involving histological changes as well as the accumulation of excessive autophagosomes and β-amyloid in their skeletal muscle fibers [29,30]. For that reason, this experimental approach could be a useful tool for better understanding of the molecular mechanisms in muscle degenerative diseases with common pathogenic phenotypes followed by the identification of autophagy signaling pathways in rats as well as in sIBM humans [31–34].
Resistance exercise is an effective method to increase muscle mass and strength [35]. Therefore, resistance training is an important therapeutic strategy that should be considered for the purpose of improving functioning, the ability to perform activities of daily living and health-related quality of life [36]. Recently, wheel running was reported to alleviate the detrimental effects of CQ on skeletal muscles in mice by repairing autophagy flux [37]. However, not only are the effects of long-term resistance training-induced autophagy in muscular disease yet to be fully defined, but the precise cellular function of Atg proteins in the autophagy signaling pathways remain to be explained [38]. Therefore, this is the first study to investigate whether the autophagy signaling pathways will provide a protective mechanism in the skeletal muscles of rats with sIBM induced by CQ treatment following long-term progressive resistance exercise training.
METHODS
Animal
Male Wistar Hannover rats (N = 18, 6 weeks of age) were obtained from the Central Laboratory Animal Incorporation (Seoul, Korea). These rats were housed in standard rodent cages with unrestricted access to water and food (temperature of 22°C, humidity 50–60%, ventilation- and light-controlled 12h to 12h light-dark cycle). The body weight of the rats was measured five times a week at the same time. The adaptation period was set for 2 weeks, after which the 8 weeks old rats were randomly assigned into three groups: 1) Control (Sham, n = 6), 2) Chloroquine (CQ, n = 6), 3) Chloroquine + Exercise (CE, n = 6).
Chloroquine-induced myopathy
The sIBM-induced rat models were treated with an intraperitoneal injection of chloroquine diphosphate salt (Sigma-Aldrich, C6628, USA) at a dosage of 50 mg/kg body weight for 5 days a week for a total of 16 weeks [31,32].
Long-term effects of resistance exercise training
The resistance training protocol for muscle disease rat models has not been examined so far. Thus, we aimed to establish a protocol applicable to subjects with muscle disease in our previous test. Referencing existing protocols [39–42], our modified method was accomplished successfully. The workout equipment consisting of climbing ladders (20 × 115 cm) with 4 cm grids inclined at 85 degrees were made in our laboratory. The rats in the CE group were trained 3 days/week (Monday, Wednesday, and Friday) for 8 weeks, for a total of 24 training sessions. In the first week, the CE group was familiarized with the ladder through voluntary climbing from the bottom to the top of the ladder. After familiarization, the resistance training began using lead weights attached to the base of the tail. To evaluate the 1 RM of CE group rats, a carrying load of 50% of the rat’s body weight was attached to each rat’s tail and gradually incremented until they reached their maximum carrying load. After determining the 1 RM, the CE group climbed the ladder with 50% (1st–2nd time), 75% (3rd–4th time), and 100% (5th–6th time) of the previous maximal carrying load. The resistance training consisted of 10 repetitions per training session, and when they reached the top of the ladder, they were allowed to recover in the resting area for 2 min. If a rat was able to climb the ladder with these loads, additional weights were placed in 50mL conical tubes at 30 g increments for each subsequent climb.
Tissue extraction
At the end of the experimental period, all animals in each group were acutely anesthetized with pentobarbital sodium (40 mg/kg i.p). After reaching a surgical plane of anesthesia, soleus muscles were removed, frozen immediately in liquid nitrogen and stored at −80°C for subsequent analyses. We assumed that the effects of CQ were not the same for each muscle fiber type. Based upon CQ-induced myopathy studies, muscle atrophy occurred predominantly in type I muscles and unusual autophagosomes were accumulated [43,44]. Therefore, this study focused exclusively on the slow-twitch soleus muscle as the representative skeletal muscle.
Western blot analysis
For protein analysis, the soleus muscles were homogenized with RIPA buffer (Biosesang, R2002, Korea) containing a Halt Protease and Phosphatase inhibitor cocktail (Thermoscientific, 78446, USA) using a homogenizer (Automill, Tokken Co., Japan). After incubation for 30 min on ice, centrifugation was conducted at 14,000 RCF for 30 min at 4°C. The resulting supernatant was collected, and protein content was assessed by the BCA method (Pierce BCA Protein Assay Kit, 23225, USA). For the running step, 40 μg of each protein was separated by SDS-PAGE with 8~15% gels for 1 hour and 30min at 80 Volts due to the molecular size of the target proteins and the proteins were transferred to PVDF membranes for 1 hour at 100 Volts. Non-specific sites were blocked for 1 hour at room temperature in the blocking solution (5% w/v BSA, 1X TBS, and 0.1% Tween-20) and then the membranes were incubated overnight at 4°C with primary antibodies. The following antibodies were used: Beclin-1 (3738, 1:1000), Atg7 (2631, 1:1000), BNIP3 (3769, 1:1000), p62 (5114, 1:1000), and LC3 (4108, 1:1000) from Cell Signaling; LAMP-2 (sc-8100, 1:200) from Santa Cruz Biotechnology; Cathepsin L (ab133641, 1:1000) from abcam. In addition, following incubation with primary antibodies, the membranes were washed extensively with 1X TBS-T for 30 min and then incubated with secondary antibodies HRP-conjugated goat anti-rabbit (1148960, 1:5000), HRP-conjugated rabbit anti-goat (811620, 1:2000) from Invitrogen, and HRP-conjugated goat anti-mouse (SC-2005, 1:5000) from Santa Cruz. After repeating the washing steps three times for 30 min at room temperature, immunoreactivity was detected with luminataTM Forte Western HRP Substrate (WBLUF 0100, Millipore, USA) in accordance with the manufacturer’s instructions. The digital images were acquired using a ChemiDoc XRS System (Bio-Rad, USA). Band intensities were quantified by densitometric analysis using Image Lab Software (Bio-Rad, USA) and values were normalized to a-tubulin for the loading control (sc-5286, Santa Cruz Biotechnology).
Data analysis
All data analyses were conducted with SPSS 18.0 software (SPSS, Chicago, IL, USA). The results are presented as means ± SE. Comparisons between groups for each dependent variable were made by a one-way ANOVA. If statistical significance was found among groups, Tukey’s honestly significantly different tests were performed for the post hoc analysis. Significance levels were established at α< 0.05.
RESULTS
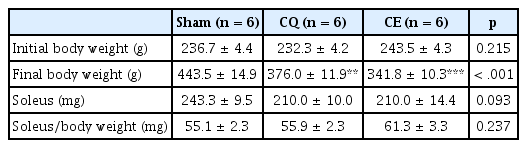
Comparisons of body weight, muscle weight and relative weight of muscle
The initial body weights among the groups were not significantly different at the early experimental stage, which established homogeneity among the groups (Table 1). In contrast, the final body weights (Sham: 443.5 ± 14.9 g, CQ: 376.0 ± 11.9 g, CE: 341.8 ± 10.3 g) were significantly different in CQ (p < 0.01) and CE (p < 0.001) compared to Sham. Thus, it was necessary to normalize muscle mass for differences in body weight as relative muscle weight. As a result, we evaluated the soleus muscle weight and the relative weight of the muscle. However, there was no significant difference between soleus muscle weight and relative soleus muscle weight among groups.
Comparisons of body weight, muscle weight, and relative muscle weight among three groups during 18 weeks
Increased maximal carrying load per resistance training session
All rats in the CE group successfully completed the entire eight weeks of resistance training. The training protocols were measured by their ability to carry progressively heavier loads. After 24 training sessions, the rats in the CE group significantly increased their maximal carrying capacity from baseline to completion by 246% (p < 0.001, Fig. 1).

Increased maximal carrying capacity per resistance training sessions during 8 weeks.
Over the course of 24 training sessions (3 times/week, total 8 weeks) the rat’s maximal carrying capacity increased 246% from baseline (315.0 g ± 20.5) to the end of the training period (776.0 g ± 41.7). Results are represented as means ± SE (n = 6).
Comparisons of autophagosome formation related to protein expression in soleus muscles
We examined the activation of the autophagy signaling pathway that begins with the formation of autophagosomes and involves a series of cascades. As a marker for the initiation of autophagosome formation, Beclin-1 protein levels were significantly increased in CQ compared to Sham (p < 0.05, Fig. 2A–B). However, the amount of elongation and formation markers for the autophagosome Atg7 protein were not significantly different among the groups, although Atg7 expression showed a decreasing tendency in CQ compared to Sham (Fig. 2A, 2C). In addition, as an inducer of mitochondrial autophagy downstream of the transcription factor FoxO3a, BNIP3 protein levels significantly decreased in both CQ (p < 0.01) and CE (p < 0.05) compared to Sham (Fig. 2A, 2D). To investigate the autophagy flux, we measured the protein levels of p62 and LC3-II as well as the LC3-II/I ratio. One of the best characterized factors of selective autophagy, p62 is an adaptor of autophagosome formation and the protein was overexpressed in CQ compared to Sham, while p62 levels in CE were significantly decreased compared to CQ (p < 0.001, Fig. 2A, 2E). Interestingly, LC3-II and the LC3 II/I ratio, which are key markers of autophagy, were not significantly different among the groups (Fig. 2A, 2F–G).
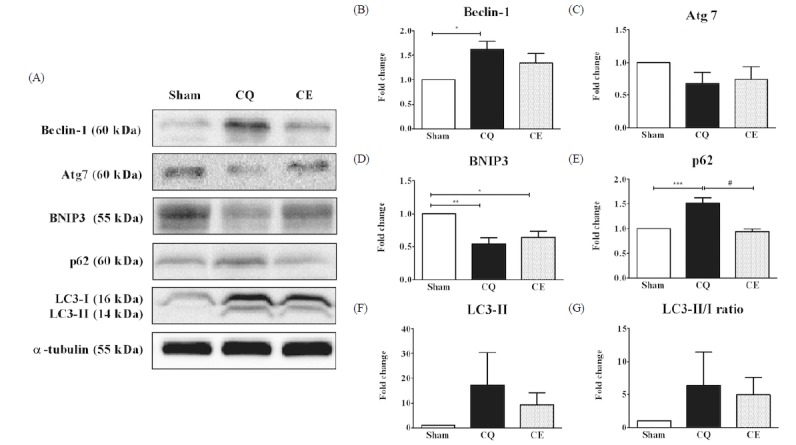
Protein levels of Beclin-1, Atg7, BNIP3, p62, LC3-II and LC3-II/I ratio in soleus muscles were analyzed as an essential marker involved in processes for the autophagosome formation. A) Representative western blot images of Beclin-1, Atg7, BNIP3, p62, LC3-II, LC3 II/I ratio and α-tubulin among groups. B) Quantification of Beclin-1 protein. C) Quantification of Atg7 protein. D) Quantification of BNIP3 protein. E) Quantification of p62 protein. F) Quantification of LC3-II protein. G) Quantification of LC3-II/I protein ratio. Protein expression comparisons were performed after normalization to α-tubulin. Results are represented as means ± SE (n = 6 rats/group). * p < 0.05, ** p < 0.01, *** p < 0.001 vs. Sham; # p < 0.001 vs. CE.
Comparisons of autophagosomal-lysosome degradation related to protein expression in soleus muscles
In the final step of autophagy, which involves the fusion of autophagosomes with lysosomes, LAMP-2 (Lysosome-associated membrane protein-2) and Cathepsin L proteins are characterized by an autophagosomal-lysosome degradation system. Specifically, LAMP-2 protein levels were significantly decreased in CQ (p < 0.01) and CE (p < 0.05) groups compared to Sham (Fig. 3A–B). However, Cathepsin L protein levels were not significantly different among the groups (p > 0.05), although the protein seemed to be downregulated in both CQ and CE groups compared to Sham (Fig. 3A, 3C).

Protein levels of LAMP-2 and Cathepsin L in soleus muscles were analyzed as a receptor at the lysosomal membrane and a marker of proteolytic capacity of lysosome, respectively. A) Representative western blot images of LAMP-2 and Cathepsin L among groups. Protein expression comparisons were performed after normalization to α-tubulin. B) Quantification of LAMP-2 protein. C) Quantification of Cathepsin L protein. Results are represented as means ± SE (n = 6 rats/group). * p < 0.05, ** p < 0.01 vs. Sham
DISCUSSION
This study is the first to investigate whether proper regulation of autophagy signaling-related proteins will provide therapeutic strategies or muscle strength improvement in sIBM-induced animal rat models after 8 weeks of resistance training. In addition, we also investigated whether the rats gradually lost body weight including muscle mass. In fact, most of the studies in vitro have been focused on the pathogenic features for abnormality instead of physical alterations [30,45]. Thus, we measured and identified whether changes in body weight, soleus muscle weight, and relative soleus muscle weight due to muscle atrophy are characterized by clinical phenotype. In our data, the final body weights in the two CQ-treated groups were dramatically decreased (Table 1). However, our experimental values didn’t show significant differences in either the soleus muscle weight or the relative muscle weight, despite the expectation of dynamic changes in the groups. Based upon the features of severe muscle atrophy, loss of body weight in CQ-treated rats are supported as the trend for chronological changes [29]. Amazingly, a few studies have reported the accumulation of vacuolated muscle fibers and β-amyloid concurrent with autophagy activation in the soleus muscles of CQ-treated rats [32,44]. Accordingly, we suspect that the autophagy signaling pathway could contribute to muscle disease development. To confirm this hypothesis, it is necessary to examine the overall steps of autophagy.
Beclin-1 is a pro-apoptotic Bcl-2 homology domain (BH3)-only protein, unlike the members of the anti-apoptotic Bcl-2 family of proteins [46]. The mammalian ortholog of yeast Atg6/Vps30, Beclin-1, is one of the essential Atg proteins that affect autophagy induction and is associated with diverse biological as well as pathological processes [47]. In the present study, Beclin-1 protein levels were significantly increased in CQ (Fig. 2A–B). A recent study demonstrated increased expression of Beclin-1 in muscle biopsies of sIBM patients and the results indicated its function in increasing sequestrating through the double-membrane of autophagosomes as well as endosome formation [34]. Although altered Beclin-1 expression levels have been observed in multiple diseases including neurodegenerative, heart, and cancer, the role of Beclin-1 in skeletal muscle disease has not been explored yet. Thus, our finding demonstrates the expression of Beclin-1 in the skeletal muscles of CQ-induced muscle disease rat models for the first time.
Atg7 is an autophagy-related E1-like enzyme that serves as the elongation and formation marker for autophagosomes and is essential for the E2-substrate reaction of LC3-lipidation [48]. Specifically, muscle-specific deletion of the Atg7 gene resulted in severe muscle atrophy and dysfunction [26]. In our results, Atg7 protein levels were not significantly different among the groups (Fig. 2A, 2C), even though the Atg7 levels showed a decrease in both CQ and CE groups compared to Sham. In fact, it is still unexplored to the Atg7 protein in sIBM patients or CQ-treated myopathy animal rats. Therefore, we suggest that it is necessary to investigate the function of Atg7 in a variety of conditions that could lead to muscle dysfunction.
BNIP3 is a pro-apoptotic BH3 only protein that induces mitochondrial dysfunction and cell death. BNIP3 is also a potent inducer of autophagy [49]. Our results revealed that BNIP3 protein levels were significantly decreased in both CQ and CE compared to Sham (Fig. 2A, 2D). Contrary to these results, the BNIP3 level was significantly increased in CQ-treated female ICR mice [37].
The autophagy flux is a very important index that explains the rates of initiation and resolution of autophagic events for maintaining muscle mass and myofiber integrity [25]. Accordingly, we monitored the expression of p62 and LC3-II proteins as well as the LC3-II/I ratio. The p62 protein, also known as sequestosome 1 (SQSTM 1), is a shuttle protein that facilitates the delivery of polyubiquitinated protein aggregates and is a common component exposed in protein aggregation diseases through interactions with LC3 at the autophagosome [50,51]. Therefore, the accumulation of p62 protein is associated with autophagy suppression, while proper turnover of p62 regulates autophagy by preventing spontaneously formed aggregates [52]. Our data showed that the p62 levels of CE significantly decreased compared to CQ (p < 0.001), while the p62 protein levels significantly increased in CQ compared to Sham (Fig. 2A, 2E). The overexpression of p62 in the CQ group in our study corresponds with findings in sIBM muscle fibers [6]. Up-regulated p62 protein expression in CQ was observed in female ICR mice, while p62 level was down-regulated in the exercise group [37]. This study showed that wheel running exercise improved deficient autophagy activation by eliminating the detrimental effect of CQ on skeletal muscles in mice. Therefore, we suggest that progressive long-term resistance exercise training could be used as a therapeutic strategy in myopathy with muscle atrophy to improve muscle function and strength.
LC3, the mammalian homolog of yeast Atg8, is localized in autophagosome membranes and has been used as an autophagosome marker to monitor autophagy. LC3 is also easily integrated into intracellular protein aggregates [53]. Two forms of endogenous LC3 are observed, the unconjugated form of LC3-I and the conjugated form of LC3-II, followed by the conversion (LC3-I to LC3-II) of LC3 when autophagy is induced [54]. To identify the autophagy activity, we detected the amount of LC3-II, which is associated with the number of autophagosomes and degraded by lysosomal hydrolases [55]. Interestingly, our data showed that the protein levels of LC3-II and the LC3 II/I ratio were not significantly different among the groups (Fig. 2A, 2E–F). In contrast, other studies reported the overexpression of LC3-II following experimental CQ-injection in rat muscles [31,32] as well as in sIBM human muscles [34]. We attributed our results to a ceiling effect because of the long-term CQ treatment. However, the results may also be due to defective autophagy resulting from long term CQ-treatment, with an excess amount of LC3-II [56].
LAMP-2 (Lysosome associated membrane protein-2) is a critical protein that is required for fusion of the autophagosome with the lysosome [57,58]. A few studies showed that increased expression of LAMP-2 in myopathies with rimmed vacuole accumulation results in abnormal lysosome function [59–61]. Contradicting these studies, our results showed that the expression of LAMP-2 proteins decreased in both CQ-treated groups compared to Sham (Fig. 3A–B). Supporting our results, other studies have also reported that LAMP-2 deficiency can be a potent inducer of myopathies evidenced in human Danon’s disease and LAMP-2 deficient mice [62,63]. For these reasons, LAMP-2 protein is proposed to have alternative roles in various disease conditions as well as species differences. Therefore, we investigated selective disturbances in lysosomal functions from the decrease in LAMP-2 levels caused by long-term CQ-treatment.
Cathepsin L is a lysosomal protease that is present in the lysosome. It is identified as a skeletal muscle atrophy marker, playing a key role in protein turnover via lysosomal degradation corresponding to the final steps of the autophagy signaling pathways [64]. Recent investigations report that the lysosomal enzyme activities of cathepsin D and B in sIBM muscles were decreased in a manner similar to our results (Fig. 3A, 3C), whereas the protein expression levels were increased [10]. Moreover, there was evidence that muscle atrophy in calpain-3 deficient mice and cathepsin L mRNA levels decreased in the skeletal muscles [65].
CONCLUSION
Our findings demonstrate that sIBM induced by CQ treatment results in muscle degeneration via impaired autophagy and that long-term resistance exercise training promotes autophagy, thus attenuating sIBM-induced muscle degeneration caused by autophagy dysfunction. Finally, our data indicate that regular resistance training provides potential protection against sIBM by enhancing autophagy flux through the p62 protein.
ACKNOWLEDGMENT
This work was supported by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2013S1A5A8023992). All processes for experiments on living animals were approved by the Institutional Animal Care and Use Committee at Korea National Sport University (Certificate KNSU-IACUC-2013-01).