INTRODUCTION
Diabetes mellitus, a complex disorder, is a combination of metabolic disorders associated with hyperglycemia due to inadequate insulin production or insulin action [18]. Diabetes is a world-wide problem affecting approximately 300 million people. In Korea, diabetic population is gradually growing. Approximately 15,000 people die due to complications associated with diabetes every year [90]. Diabetes is classified into two groups, type 1 and type 2. Type 1 is characterized by beta-cell destruction leading to insulin deficiency due to autoantibody formation. Type 2 is characterized by insulin resistance and eventual reduced insulin secretion due to pancreatic scaring and loss of beta-cells. Type 2 diabetes, comprising 90-95% of all cases of diabetes mellitus, is one of the most lethal diseases in the world [88].
The main cause of death in type 2 diabetes is cardiovascular disease, specifically atherosclerosis [58]. Atherosclerosis is initiated by sequences of alterations in the structure and function of the vascular endothelium [2,11]. The exact etiology of vascular endothelial dysfunction is complex without well understanding. However, experimental evidences suggest that imbalance between oxidative stress and host antioxidant defense along with pro-inflammatory and anti-inflammatory factors play critical roles in early vascular endothelial dysfunction [58]. Some (but not all) previous exercise interventions have resulted in enhanced vascular endothelial function in type 2 diabetics. However, limited information is available on the most effective type of exercise training program or mechanisms responsible for the improvements seen in vascular endothelial function. Thus, this review has the following three aims: 1) to introduce presumed diabetes-specific mechanisms responsible for dysfunctional vascular endothelium; 2) to summarize and investigate current evidence of the effect of exercise training on conduit vessel endothelial function in type 2 diabetic patients; and 3) to present possible future directions to what should be further explored to expand our knowledge on this research topic.
Relaxation of vascular endothelium: a major determinant for vascular integrity
Human vasculature is composed of three layers: the endothelium (intima), smooth muscle cells (media), and surrounding elastic and connective tissues (adventitia). The vascular endothelium comprises the innermost layer of the vasculature, which directly senses changes in blood flow and interacts with hormones and neurotransmitters through various receptor-ligand complexes at its membrane, producing vasoactive agents such as nitric oxide (NO), prostacyclin (PGI) , endothelium-derived hyperpolarizing factors (EDHF) , and endothelin-1 [45,55,65]. These agents control vascular tone at the vascular smooth muscle level either through vasoconstriction or vasodilatation. The vasculature is relaxed or dilated if the effect of dilatory agents overrides that of constricting agents, such as the basal sympathetic tone and endothelin-1, whereas vasoconstriction occurs if the dilatory signals are overpowered.
Nitric oxide or NO, the most prominent vasodilatory agent, is produced by the L-arginine - endothelial nitric oxide synthase (eNOS) pathway as a byproduct. The L-citrulline in the vascular endothelium then diffuses into vascular smooth muscle cells and facilitates soluble guanylyl cyclase to convert guanosine triphosphate (GTP) to cyclic guanosine monophosphaste (cGMP), which in turn leads to calcium ion movement into the sarcoplasmic reticulum, decreasing calcium ion concentration within the cytoplasm of smooth muscle cells, thus causing the vascular smooth muscle cells lose their tonicity [19,31,37,59,78]. Similarly, PGIs are produced when cyclooxygenase (COX) uses arachidonic acid (AA) as a substrate in vascular endothelium to move into neighboring vascular smooth muscle cells, which then convert adenosine triphosphate (ATP) into cyclic adenosine monophosphate (cAMP) through activated adenylyl cyclase (AC), inducing vasodilation as a result of decreased calcium ion concentration in vascular smooth muscle cells [4,36,44].
The endothelin derived hyperpolarizing factor (EDHF) signaling pathway still remains to be elucidated although there have been many studies [1,13,24,37,52,56]. However, it is known that EDHFs are generated by the enzymatic activity of cytochrome P-450 within the golgi apparaturs in endothelial cells. Like COX, AA used as a substrate for the enzymatic activity of cytochrome P-450 eventually leads to EDHF stimulation or opening of potassium ion channels on the vascular smooth muscle cell membrane, resulting in hyperpolarization of vascular smooth muscle cells. This potassium mediated hyperpolarization reduces the duration of calcium ion channel opening, resulting in decreased calcium ion influx into vascular smooth muscle cells which leads to vascular smooth muscle relaxation [47,55,76]. Endothelin-1 (ET-1) is an endogenous constricting agent produced in vascular endothelial cells that acts as a vascular smooth muscle constricting agent after binding to ET-1 receptors (ET-A or B receptors) on the vascular smooth muscle cell membrane [46,49,70,84]. The endothelium-dependent vasoregulatory signaling pathways are illustrated in Fig. 1.
As previously mentioned, the modulation of human vasculatures is very complicated since there are numerous physiological mediators involved neural (release of norepinephrine) and humoral (angiotensin II and epinephrine) regulators which systemically influence and interact with more local regulators, such as the myogenic response and endothelium-derived relaxing factors (EDRF: NO, PGI, and EDHF) [14,30,60,80]. However, EDRFs are considered to be the most important vasoregulators because they override resting vascular tone controlled by systemic regulators and fine tuning other local mediators [60].
Nitric oxide (NO) also prevents abnormal growth of vascular smooth muscle cells and maintains optimal, healthy vascular wall structure [10,43]. Functionally, NO is the major vasodilator modulating systemic vascular resistance, whereas PGI and EDHF are less influential by providing supplementary means for vasodilatation [12]. Improved NO production along with NO bioavailability depend on the eNOS substrate L-arginine’s availability, eNOS phosphorylation and dimeric coupling, availability of eNOS cofactors (tetrahydrobipterin: BH4, heat shock protein-90: HSP-90, and calmodulin), and a balance between oxidants and antioxidants [3,20,38,53,68,69,72]. Thus, it is important to assess systemic biomarkers with NO-mediated vasodilation in order to understand the mechanisms involved in the structural and functional changes that occur in diabetes patients.
Potential mechanisms of diabetes-mediated vascular endothelial dysfunction
Unlike other tissues and cells not affected by abnormal systemic glucose concentration, vascular endothelium is very sensitive to alterations of blood glucose. Therefore, vascular endothelium is likely to be a main target of hyperglycemic damage [40]. Oxidative stress has emerged as a major player in the cause of vascular endothelial dysfunction in other chronic diseases such as diabetes as well as in normal aging. However, in diabetes, it has been recognized that hyperglycemia-induced generation of reactive oxygen species (ROS) in the vascular endothelium is the foundation of micro- and macro-vascular complications [5].
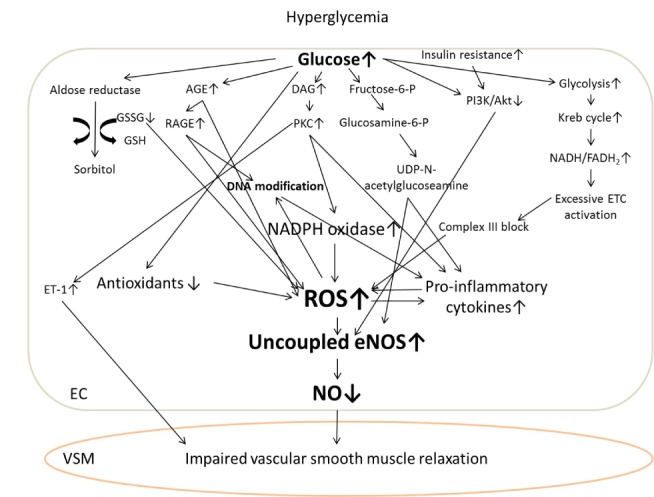
Four putative mechanisms of diabetes-mediated vascular complications have been indicated by well-designed molecular studies. Firstly, the altered role of aldose reductase through sorbitol aldose reductase pathway increases oxidative stress, thus inducing the formation of uncoupled eNOS. Unlike its normal function to convert aldehyde to inactive alcohol, aldose reductase converts glucose into sorbitol which abnormally increases intracellular glucose concentration. During this signaling process, aldose reductase uses nicotinamide adenine dinucleotide phosphate (NADPH) as a cofactor. Thus, the absolute quantity of NADPH, a major cofactor in regenerating reduced glutathione, is decreased, further augmenting intracellular oxidative stress [34,81]. Secondly, hyperglycemia-induced advanced glycation end products (AGEs) in vascular endothelium cause increased production of ROS and inflammatory cytokines as well as increased receptor (RAGE) activity and pro-atherogenic lipoprotein modification [35,86]. Thirdly, an overall increase in diacylglycerol (DAG) production leads to protein kinase C (PKC) activation. DAG, a mediator in the second messenger system, is recruited for intracellular lipid metabolism. After DAG activation, PKC is involved not only in eNOS activity, but also in a lot of gene expression and cytokine production in the vascular endothelium. Activated PKC increases ROS generation by NADPH oxidase, vascular adhesion molecules, pro-inflammatory cytokines, and growth factors, but decreases eNOS activity, resulting in a decrease of overall NO bioavailability, a major characteristic of vascular endothelial dysfunction [27,33,61,64,82]. Fourthly, fructose 6-phosphate can be converted into glucosamine 6-phosphate by glucosamine 6-phosphate transferase through the hexosamine pathway in the hyperglycemic cellular environment, instead of being converted to glyceraldehyde 3-phosphate in the glycolytic pathway. Uridine diphosphate (UDP) N-acetyl glucosamine, the cytosolic end product of the hexosamine pathway, decreases eNOS phosphorylation but increases transforming growth factor-β1 and plasminogen activator inhibitor-1 production, which in turn facilitates the pathological processes of diabetic vascular complications [25,42,71,85]. Currently, these four distinct mechanisms have not been directly associated with each other. However, they appear to have a common upstream inducer hyperglycemia and abnormally augmented ROS production along the electron transport chains (ETC) within vascular endothelial cell mitochondria [5].
According to Brownlee’s concept on the role of oxidative stress in diabetes-mediated vascular complications, hyperglycemia facilitates the rate of glycolysis and the Krebs cycle, which aberrantly generates more nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) and abnormally over-activates ETC. Once the ETC is activated, more hydrogen ions come out of the mitochondrial inner membrane which increase membrane potential [5]. To bring the membrane potential back down to normal, complex III in the ETC is blocked, electrons from coenzyme Q combined with oxygen molecules then generate more superoxide anions. However, hyperglycemia reduces both enzymatic (superoxide dismutase, catalase, and glutathione peroxidase) and non-enzymatic (vitamin E, coenzyme Q, lipoic acid, and glutathione) antioxidants through the over production of ROS [5]. Thus, the balance between oxidants and antioxidants is disrupted which becomes a main trigger of diabetic vascular complications.
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, a major ROS source in the cytosol, leads to augmented superoxide production in vascular endothelium in the presence of abnormal glucose concentrations [41]. Hyperglycemia and increased ROS also results in structural and functional abnormality of the eNOS system. Insulin resistance, another major symptom of diabetes, inhibits eNOS phosphorylation by down-regulating phosphoinositide 3-kinase and PI3K/Akt pathway in vascular endothelium, decreasing NO production and thus its bioavailability [87]. The increased generation of reactive nitrogen species (i.e., peroxynitrite) within the vascular endothelium in diabetics could be also due to hyperglycemia. These reactive nitrogen species help facilitate lipid peroxidation, nitrotyrosine production, and DNA modification, leading to cardiovascular complications such as atherosclerosis [89]. Activated vascular adhesion molecules pooled into vascular endothelium also contribute to atherosclerotic plaque development in diabetics [17]. Putative pathways related to hyperglycemia-induced vascular endothelial dysfunction are illustrated in Figure 2.
Assessment of conduit artery endothelial function: Flow-mediated dilation (FMD)
Flow mediated dilation or FMD is the non-invasive gold-standard way to assess NO-mediated vascular endothelial function in response to a physiological stimulus as seen in reactive hyperemia. Since this methodological approach was introduced by Dr. Celermajer in the Lancet Journal in 1992, many cardiovascular physiologists who work on human subjects have employed and further developed this methodology [6,11,16,21,26,29,75,77]. Unlike other methods such as venous occlusion plethysmography which indirectly assesses changes in local smaller vessel blood flow as segmental volume changes of a limb in response to local invasive pharmacological stimulus (i.e., acetylcholine), FMD directly measures changes in larger conduit vessel diameter in response to reactive hyperemia, which occurs after 5 minutes of local limb blood flow occlusion [26]. FMD determines the dilatory capacity of larger vessels by determining the difference between the resting diameter and the diameter during peak reactive hyperemia. FMD also has a strong correlation with invasive coronary epicardial vasoreactivity when evaluating conduit artery function. It has benefits in assessing other important functional parameters such as blood flow, velocity and vascular conductance [16,29]. However, FMD requires experienced operator to perform it accurately. In addition, it needs unified guidelines and standardizations in order to have validity when comparing results to other studies. Recently, Dr. Dan Green’s group provided a methodological guideline to assess FMD [77]. Although there are differences in operators, equipment, devices, analysis tools, and software between laboratories, unified technical guidelines will help researchers to accurately measure and evaluate FMD, and to compare the outcomes found in different laboratories. Most importantly, vascular smooth muscle responsiveness to a nitric oxide donor (i.e., nitroglycerin) needs to be tested if FMD method is used in order to ensure that results are coming specifically in an endothelium-dependent manner. In many studies, the outcomes of FMD have been supported by assays of numerous systemic biomarkers. Since Dr. Feng and Dr. Colombo introduced and developed a state-of-the-art methodological approach which collects vascular endothelial cells by inserting guide wires into artery or vein via catheterization and evaluates local endothelial cell protein expressions using immunofluorescence, such method has provided an abundant amount of scientific evidence to understand the mechanisms associated with vascular endothelial function [9,22,23,28,32, 39,62,63,73,74]. Thus, assessing both systemic and local biomarkers with FMD would be a great methodological approach to understand the mechanisms involved in any functional changes.
Aerobic exercise training and vascular endothelial function in type 2 diabetic patients
Although there has been a substantial effort to promote vascular health in type 2 diabetic patients, only a few randomized controlled trials have tested the effect of aerobic exercise training on vascular endothelial function in type 2 diabetic patients with inconclusive and controversial results [54]. Some experimental evidence demonstrated a positive influence of regular physical activity and aerobic exercise on the improvement of the vascular endothelial function through systemic inflammatory biomarkers in diabetics. However, other studies indicated that regular aerobic exercise did not enhance the impaired endothelial function or systemic biomarkers in type 2 diabetic patients [15,48,50,51,57,66,88]. Although there has been a lot of attention on the effect of aerobic exercise on large conduit artery health mainly assessed by FMD, only a couple of studies have evaluated the effect of aerobic exercise training specifically on conduit artery endothelial function in type 2 diabetics [48,50]. According to the recent collaborative statement by the American College of Sports Medicine (ACSM) and the American Diabetes Association (ADA), either aerobic or combined exercise (aerobic & resistance training) is recommended to prevent diabetes and the associated complications specifically to improve diabetic vascular health [8]. However, the recommended guidelines for aerobic exercise training are still nonspecific without specific aerobic exercise regimen aimed specifically towards helping improve vascular endothelial function in type 2 diabetic patients. This is partly due to the fact the mechanisms of the beneficial effects that aerobic exercise training has on diabetic vascular complications have not yet been fully elucidated. Recently, Dr. Wisloff and his colleagues have demonstrated that 16 weeks of high-intensive interval training was greatly superior to moderate continuous exercise on the improvement of the vascular endothelial function, insulin signaling, and overall blood glucose in metabolic syndrome patients, including patients with prediabetes [79]. The proposed effects of high-intensity interval training has caused patients and health care providers alike to become skeptical on its benefits when considering its potential risks and secondary detrimental effects it might have on individuals who are not physically fit. However, it has been recently demonstrated that high-intensity interval training can be safely employed in patients of high risk groups such as those with heart failure, metabolic syndrome, coronary artery disease, or hypertension. High-intensity interval training has been tested to over 2000 hours of intervention without any negative events [67,79,83]. Accordingly, future studies should demonstrate the benefit of high-intensity interval training on vascular endothelial function in type 2 diabetic patients. Furthermore, the optimal training modality, intensity, frequency, and duration to improve impaired and dysfunctional diabetic vasculature should be emphasized.
CONCLUSION
Hyperglycemia and insulin resistance are representative characteristics of type 2 diabetes. Vascular endothelial cells are highly sensitive to hyperglycemia along with insulin resistance. Nitric oxide (NO) is a major constituent necessary to maintain vascular integrity and dilatory capacity. FMD is the gold standard measure to assess NO-mediated vascular endothelial function. Diabetes-mediated vascular complications have their own potential mechanisms. Increased ROS generation of endothelial mitochondria has been implicated as a common precursor mechanism in the formation of diabetic vascular complications. Even though some studies have explored the effects of exercise on the human vascular endothelial function and related mechanisms, the outcomes in these studies were not consistent. The mechanistic understanding was limited to some systemic inflammatory biomarkers. Thus, future studies need to demonstrate the optimal exercise regimen for impaired diabetic vasculature and to directly investigate related molecular and biochemical mechanisms involved in the induction and prevention of endothelial injury within collected vascular endothelial cells in addition to traditional systemic biomarkers.